01研究背景
肝细胞癌(HCC)是全球第四大癌症死亡原因,泛素-蛋白酶体系统、NF-κB通路、自噬等细胞信号通路都参与了HCC的发生发展。TRAF2作为衔接蛋白和泛素E3连接酶,在介导TNFα-NFκB信号通路中发挥重要作用,并在多种人类癌症中表达失调,但TRAF2是否调节肝癌细胞的生长尚不清楚。
2023年, 浙江大学医学院第一附属医院郑敏、赵永超团队在“Cell Death and Differentiation (IF 12.067)”上发表文章“The TRAF2-p62 axis promotes proliferation and survival of liver cancer by activating mTORC1 pathway”,发现TRAF2通过促进p62多泛素化来增强其与Raptor-RagC的结合,导致mTORC1激活,进而促进肝癌的发展,表明TRAF2是肝癌治疗的潜在靶点。
· 维真助力 ·
维真生物提供文中所用慢病毒载体:pLent-shRNA-p62
02部分结果展示
1. TRAF2负调控p62蛋白水平和自噬通量
作者发现HCC细胞系和HCC临床组织中TRAF2 mRNA水平高于正常肝脏细胞和邻近非肿瘤组织,同时还与HCC患者预后不良高度相关。为了探索TRAF2促进肝癌进展的机制,作者通过串联亲和纯化和质谱分析,发现p62是TRAF2结合的候选蛋白。结构域分析发现,TRAF2的TRAF结构域和p62的TB结构域介导了TRAF2-p62的结合。为了探讨TRAF2对p62的影响,作者研究了TRAF2是否影响HCC细胞系中p62蛋白的表达,发现敲低TRAF2显著增加了HepG2和Huh7细胞中的p62蛋白水平,TRAF2过表达以剂量依赖的方式降低内源性p62水平。p62参与自噬调控,作者进一步研究了TRAF2对自噬的影响,发现TRAF2敲低会破坏HCC细胞的自噬通量。
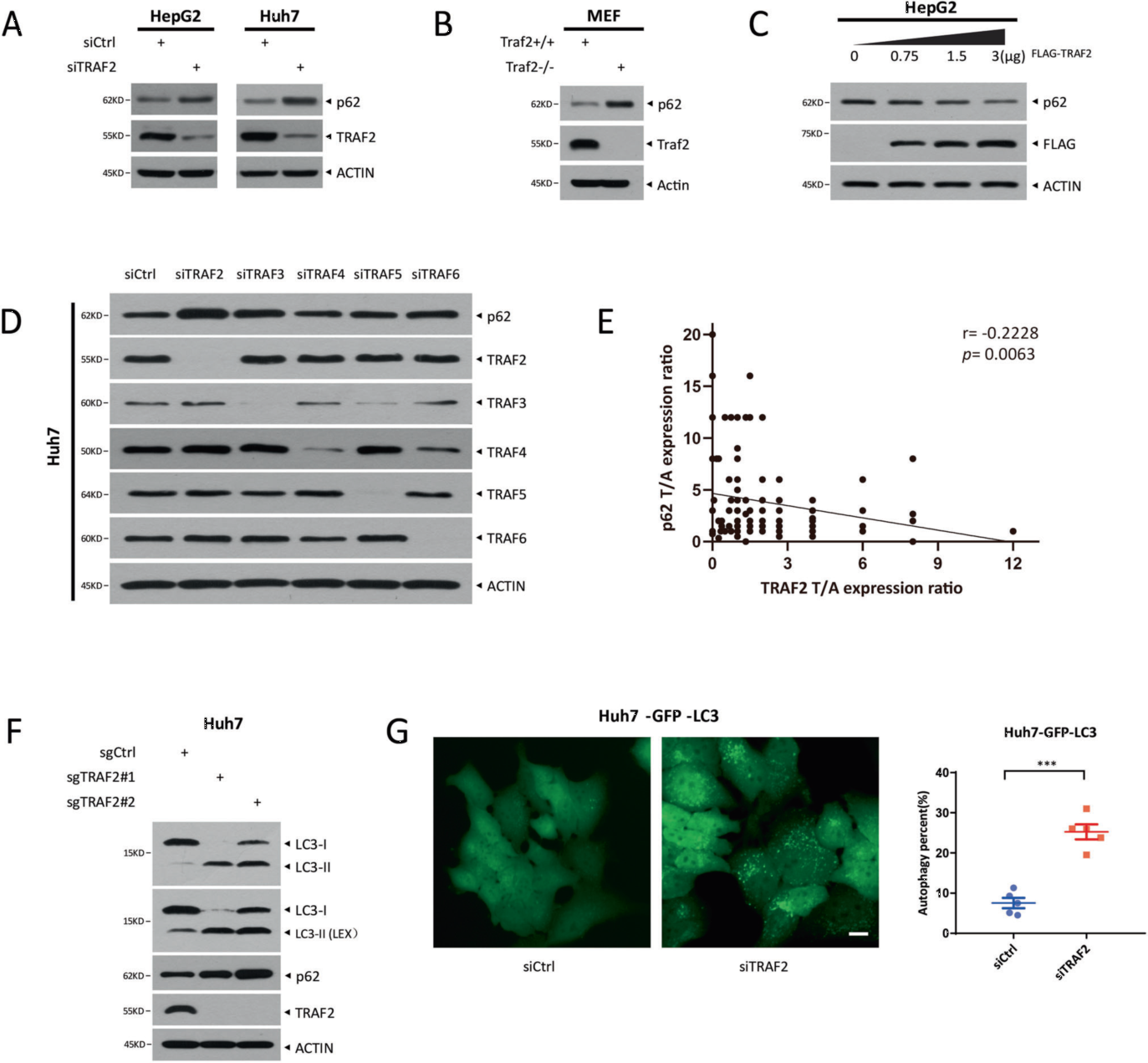
图1. TRAF2负调控p62蛋白水平和自噬
2. TRAF2促进p62的k63连接的多泛素化和溶酶体依赖性降解
由于TRAF2与p62相互作用并负向调节p62蛋白水平,作者假设TRAF2介导p62的降解。为了验证这一假设,作者先研究了TRAF2对p62蛋白半衰期的影响,发现TRAF2敲低后p62的半衰期延长。之后作者研究了p62是否受到TRAF2诱导的多泛素化,发现TRAF2显著促进p62多泛素化,进一步研究发现TRAF2促进p62在420赖氨酸残基上的k63连接的多泛素化,且p62的降解是通过溶酶体介导的,而不是蛋白酶体系统。
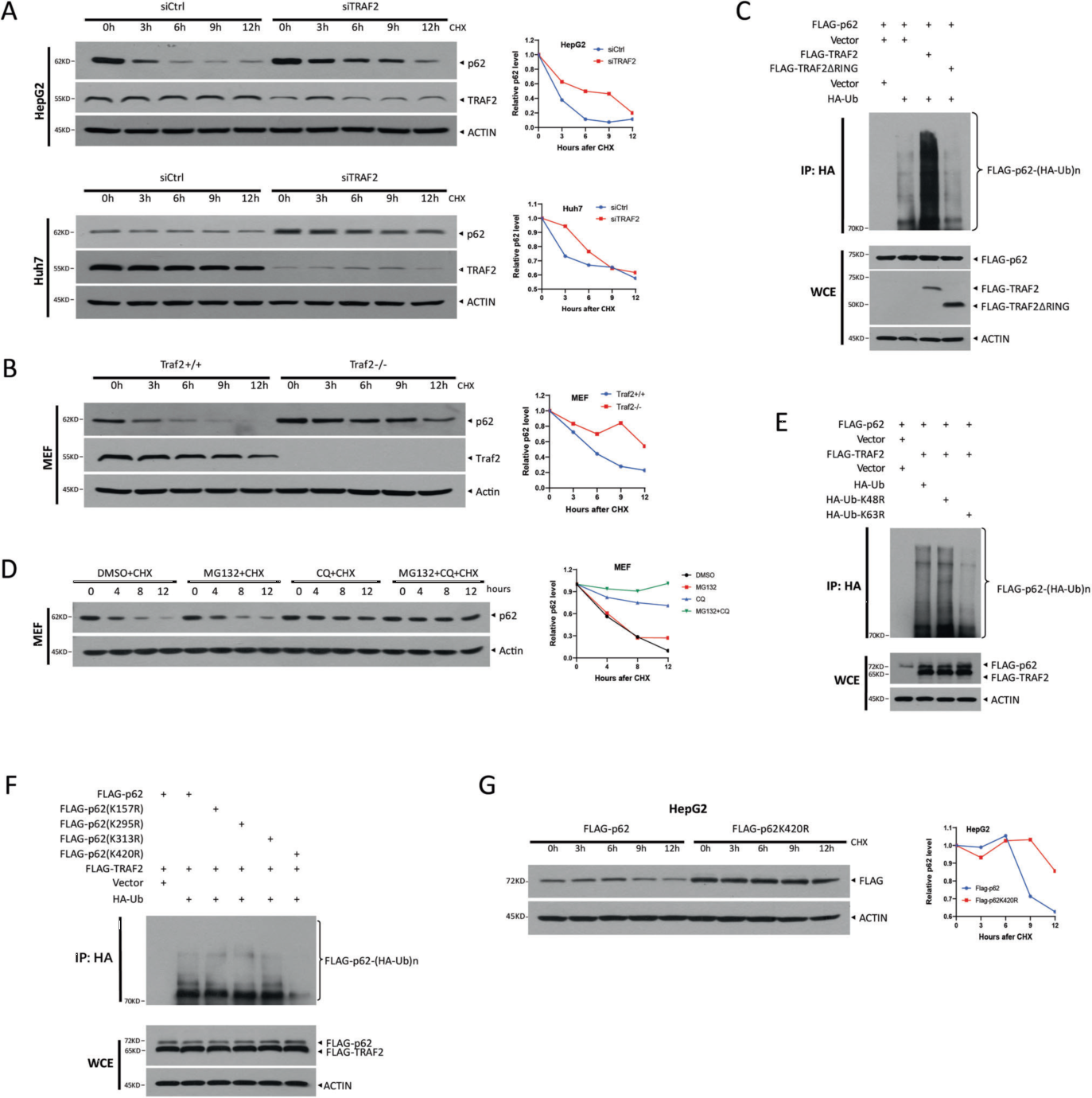
图2. TRAF2促进p62的k63连锁多泛素化和溶酶体依赖性降解
3. TRAF2在体外和体内以p62依赖的方式促进肿瘤生长
由于p62是TRAF2的底物,作者接下来研究了p62在TRAF2敲除肝癌细胞中的作用机制,发现在TRAF2缺失的细胞中,p62的敲低促进了肝癌细胞的生长和存活,表明p62起抑制生长的作用。之后作者用小鼠异种移植肿瘤模型验证了这一结果,将表达p62 shRNA的慢病毒转染到TRAF2缺失的Huh7细胞中,扩增后接种裸鼠,发现p62敲低显著逆转TRAF2敲低对肿瘤生长的抑制作用,肿瘤体积和重量增加。进一步研究发现,p62敲低通过激活mTORC1活性,在体外和体内挽救了TRAF2敲低对肿瘤生长抑制。
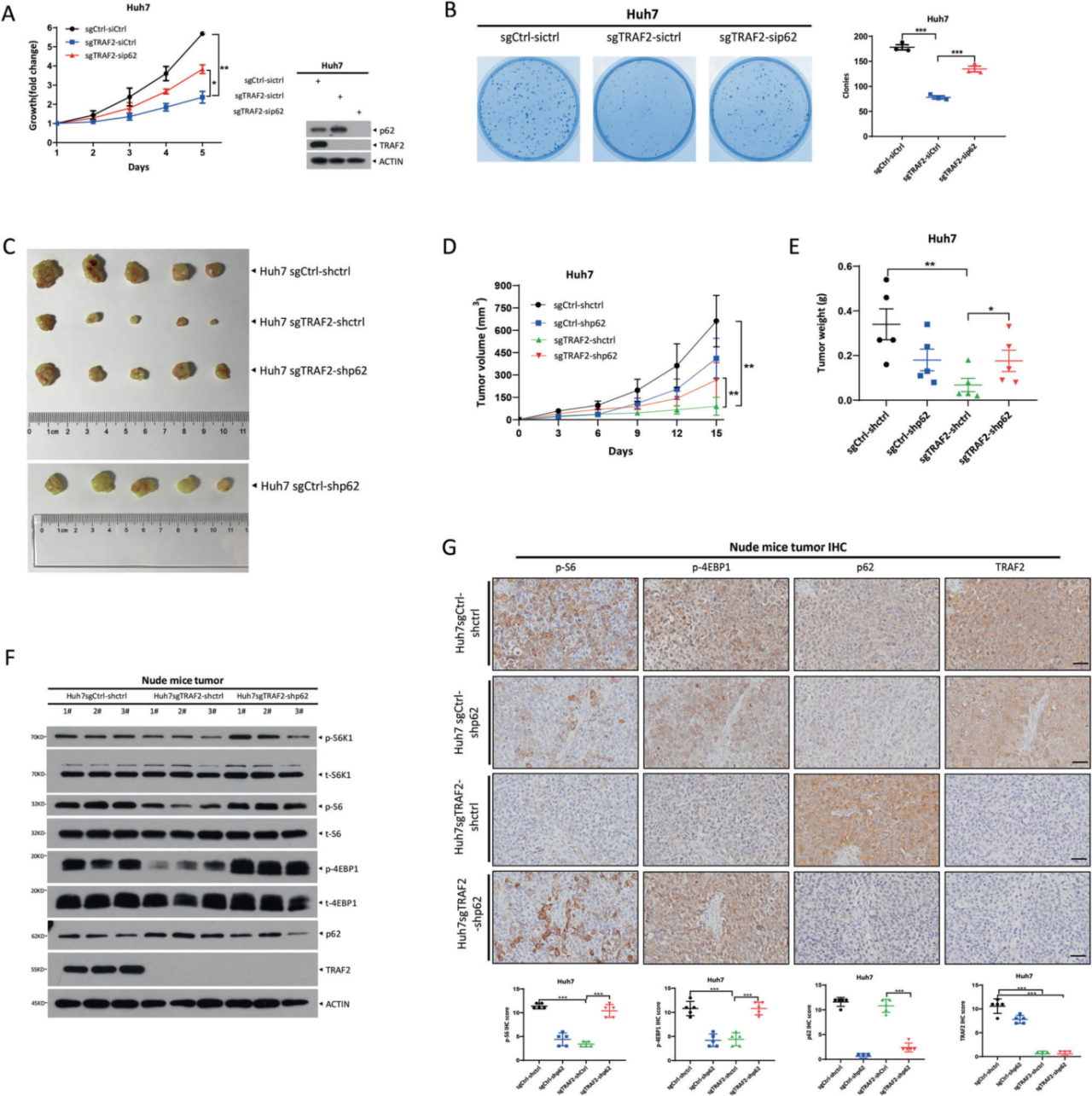
图3. TRAF2在体外和体内以p62依赖的方式促进肿瘤生长
4. TRAF2泛素化p62促进mTOR激活
由于p62敲低挽救了TRAF2敲除对生长的抑制,作者假设泛素化的p62可能是调控HCC生长的主要作用形式。为了验证这一假设,作者将p62与其泛素死亡突变体重新转染到p62缺失的Huh7细胞中,发现p62能显著促进细胞和裸鼠异种移植模型的生长,并重新激活了肿瘤组织中的mTORC1活性,但p62泛素死亡突变体无上述功能。之后作者研究了p62泛素化调控mTORC1活性的机制,发现与p62相比,突变体抑制Raptor和RagC的结合,并导致mTOR与LAMP2的共定位减少,表明mTORC1的再激活减少。综上所述,泛素化p62似乎是mTORC1激活的原因,mTORC1激活进一步促进了肝癌细胞的生长。
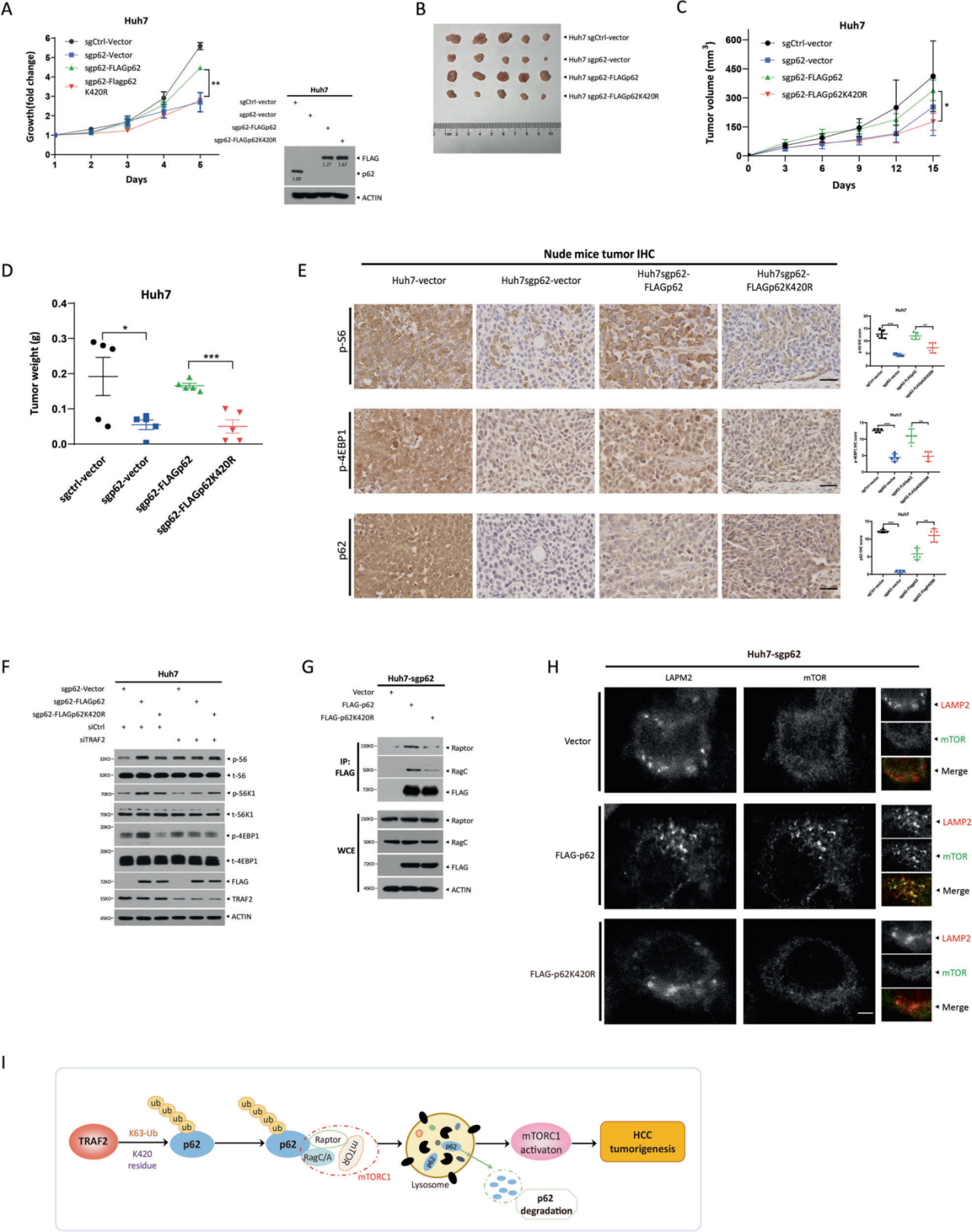
图4. TRAF2泛素化p62促进mTOR激活
03实验结论
本研究发现TRAF2促进肝癌细胞生长和存活。从机制上讲,TRAF2通过促进p62多泛素化来增强其与Raptor-RagC的结合,从而导致mTORC1激活,进而促进了肝癌细胞的生长,表明TRAF2是肝癌治疗的潜在靶点。