
肝星状细胞(HSCs)是一种非实质肝周细胞,因其是肝损伤的原发性纤维化细胞类型备受关注。HSCs被认为是一种可塑的细胞类型,可以调节肝脏生长、免疫和炎症,以及能量和营养稳态,这些功能依赖于代谢的多样性和能量消耗的严格调控。然而HSCs通过其可塑性调节多种生理和病理反应的机制仍不清楚。HSCs是一个被忽视的决定免疫代谢的因素,在支持肝功能和免疫应答损失中,细胞从静止状态激活或转分化为增殖的、运动的肌成纤维细胞,分泌细胞外基质,需要快速适应以满足更高的能量需求,这些适应包括中心碳代谢的编程、线粒体数量和活性的增强、内质网应激,以及通过储存在细胞质液滴中的视黄酯自噬依赖性水解而释放游离脂肪酸。本文揭示了HSCs的代谢调节如何在健康和疾病中发挥其功能。

背景
星状细胞(以前称为脂肪细胞、脂肪储存细胞、窦周细胞或Ito细胞)来源于隔膜横隔间充质,间充质来源的间皮和间皮细胞从胚胎肝脏表面向内迁移,产生HSCs和其他血管周围间充质细胞。正常的肝脏中,HSCs在核周液滴中储存维生素A作为视黄酯,这些细胞是人体维甲酸储存的主要仓库。
单细胞转录方法在表征HSCs方面有非常广泛的应用。在肝损伤期间,HSCs转分化,或“激活”成增殖的、纤维化的肌成纤维细胞(MFBs),这些细胞获得一系列共同维持损伤和纤维化的特征(Figure 1),由于肝损伤通过凋亡或逆转为“失活”表型来减少HSC衍生的MFBs数量。这些失活细胞具有独特的表观遗传特征,并且在未来HSCs再损伤时更容易重新激活。
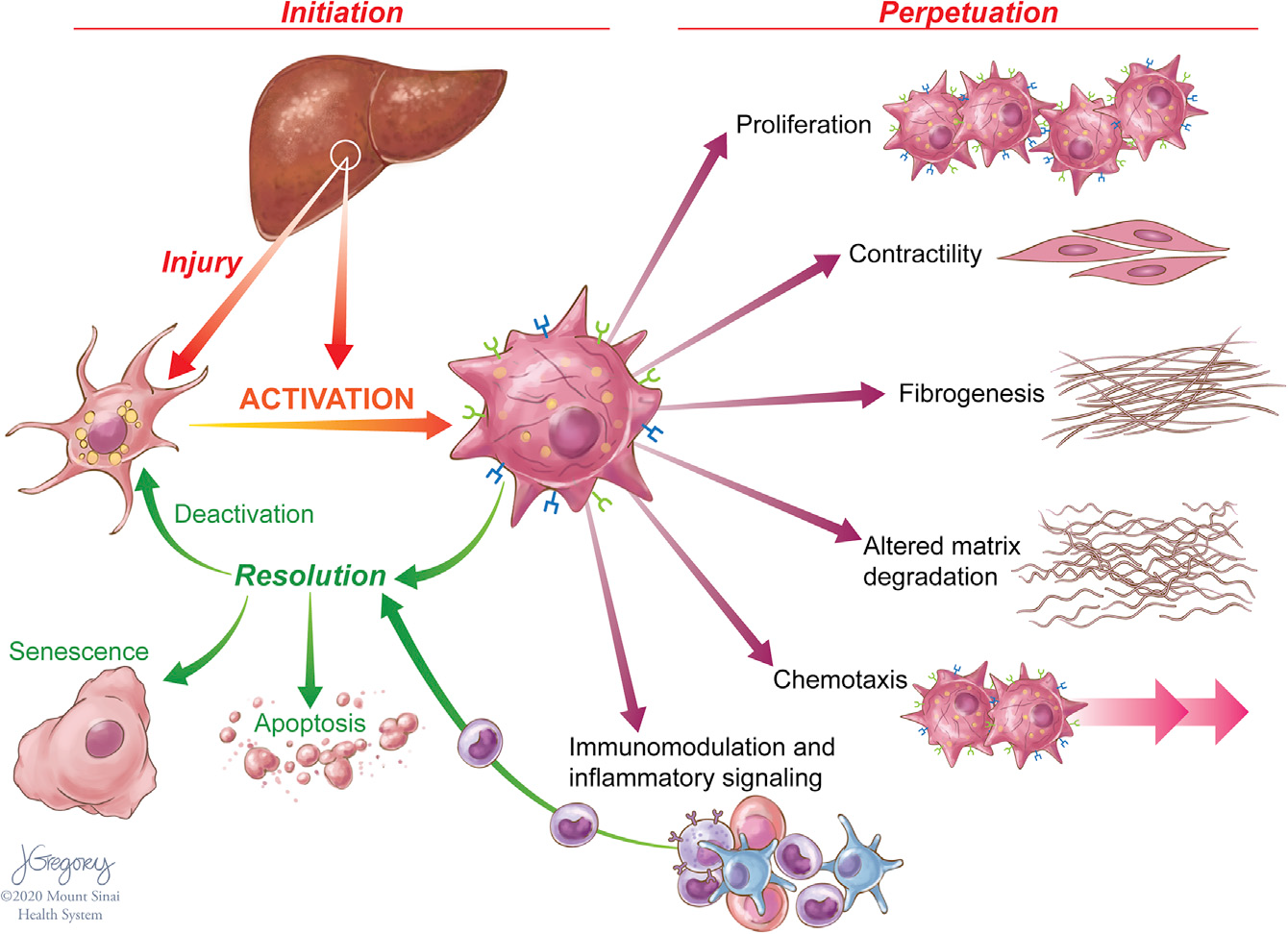
Figure 1. Features of HSC Activation and Resolution
慢性肝损伤可促进HSCs的持续激活,导致细胞外基质(ECM)蛋白的积累,从而破坏肝脏的结构及其功能。HSC的激活是由许多信号触发的,这些信号是细胞损伤的标志,包括浸润免疫细胞产生的促炎细胞因子、肝细胞凋亡小体、内皮细胞介导的生长因子激活和活性氧负担增加。细胞对损伤的反应通过一系列旁分泌和自分泌环增强,包括转化生长因子β1(TGF-β1)和结缔组织生长因子(CTGF)等纤维化信号。激活HSCs需要能量来支持细胞增殖,分泌ECM、蛋白酶和细胞因子,并向细胞损伤区域迁移的能力。这些细胞利用了许多具有很高能量需求、类似于癌细胞的代谢途径。
HSC生物能学BIOENERGETICS
碳水代谢-糖酵解和糖异生
HSC中心碳代谢重编程在细胞活化中起着重要作用,因为它们上调糖酵解以满足转分化为肌成纤维细胞表型的能量需求(Figure 2)。HSCs葡萄糖稳态调节的研究几乎完全集中在培养的细胞,因此未来的研究将需要在体内疾病模型中验证这些发现。与静息的HSCs相比,活化的人HSCs(aHSCs)和培养活化的大鼠HSCs提高了葡萄糖利用率,原代培养激活细胞增强了葡萄糖转运能力和糖酵解活性、葡萄糖转运体GLUT1、GLUT2和GLUT4在原代培养激活永生性的大鼠HSCs中表达,并可在这些条件下通过高细胞外葡萄糖或嘌呤能信号进行调节,此外GLUT1在癌细胞中也有很高的表达。
在永生化的人和原代小鼠HSCs中,与葡萄糖细胞内过程相关基因的mRNA表达也增加,如己糖激酶2(HK2)、果糖-2,6-双磷酸酶-3(PFKFB3)和丙酮酸激酶(PK)。这些酶诱导的糖酵解增强伴随着糖异生相关基因的下调,包括磷酸烯醇丙酮酸羧激酶1(PCK1)和果糖双磷酸酶1(FBP1)。培养中HSCs的糖酵解增加伴随着中心碳代谢产物远离柠檬酸循环。活化的HSCs增加了丙酮酸脱氢酶激酶3(PDK3)的表达,PDK3抑制丙酮酸向乙酰辅酶A的转化,进而乳酸积累,类似于癌细胞中的Warburg效应。丙酮酸激酶M2(PKM2)可能是aHSCs和癌细胞之间独特的联系,它在两种细胞类型中被诱导以促进需氧糖酵解。
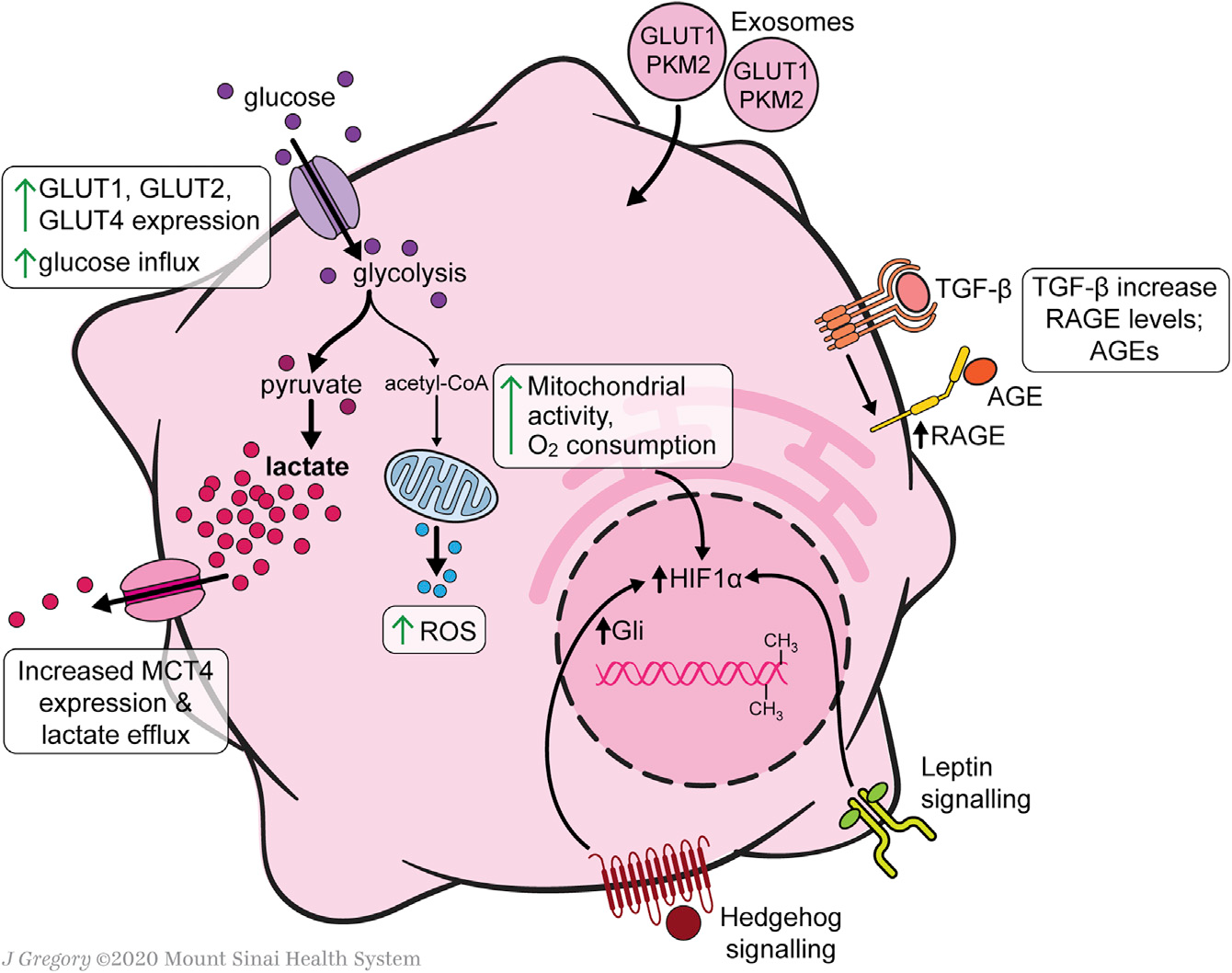
Figure 2. Glucose and Mitochondrial Metabolism in Activated HSCs
乳酸在HSC代谢中直接参与激活和MFB表型的永久化。尽管乳酸输出泵单羧酸转运体4(MCT4)的表达上调,但aHSCs细胞内乳酸水平仍升高。阻断乳酸在细胞内积累可以抑制增殖,抑制与MFB特征相关的基因,并诱导脂质积累和脂生成基因。外泌体为快速诱导糖酵解提供了一种机制,以支持从静息的HSCs(qHSCs)到aHSCs的代谢重编程,从而实现基质细胞损伤反应的同步。缺氧诱导因子1-α(HIF-1α)信号增强了外泌体的产生,进而刺激了缺氧和炎症条件下HSC的激活。此外,表观遗传调控也有助于HSCs葡萄糖代谢的转化。
糖基化
除了碳水化合物分解代谢的变化外,aHSCs还会改变蛋白质糖基化谱。aHSCs细胞表面糖基含有有利于加连蛋白-1(Gal-1)结合的葡聚糖。与qHSCs相比,aHSCs中上调的Gal-1与神经素-1(NRP-1)结合,NRP-1是一种诱导血管再生、血管通透和伤口愈合的受体。Gal-1和NRP-1的相互作用诱导了TGF-β1和PDGF样信号级联。
高胰岛素血症和高血糖
高胰岛素血症和高血糖是代谢综合征的关键特征,可激活培养的HSCs,但其在NAFLD和NASH状态下对HSC激活的具体贡献尚不清楚。在高糖培养基中培养的原代小鼠HSCs增加了I型胶原的产生,这是由SMAD3转录因子介导的。高糖暴露导致Stat4(PIAS4)蛋白抑制剂的HSC上调,抑制SIRT1的转录。减少SIRT1导致SMAD3的赖氨酸高乙酰化,从而增加SMAD3对其促纤维化DNA结合靶点的亲和力。然而高血糖和高胰岛素血症与HSC激活之间的联系也是基于培养研究,缺乏体内研究。
增强的糖基化终产物(AGEs)是由蛋白质和糖之间的非酶反应产生的,也能激活HSCs。研究表明单独AGEs和高血糖可能不足以诱导纤维化,但在组织损伤的情况下,它们可能会放大由其他因素驱动的纤维化发生。
线粒体代谢
尽管糖酵解优先增加,维持肌成纤维细胞样表型仍然需要氧化磷酸化的主要能量贡献,比如细胞激活过程中线粒体数量和活性的增加。而线粒体代谢的变化可能不局限于ATP的产生,还有ROS的作用。培养激活的HSCs具有丰富的线粒体,这一数量的增加伴随着线粒体活性的提高,如耗氧率、线粒体膜电位和F1-Fo ATP酶的表达,线粒体解偶联可抑制体外HSCs的激活,也使ATP的产量减少了30%。尽管糖酵解上调,ATP产生的一个重要和必要的部分仍然是通过氧化磷酸化。高胰岛素血症小鼠中HSCs的线粒体体积增强,类似于在培养基中激活的HSCs的变化。
来自其他组织的成纤维细胞群体提供了更多的参考。例如,肾成纤维细胞中,TGF-β1活性通过抑制电子传递链成员线粒体蛋白复合物III而增加ROS的产生。心脏成纤维细胞中,TGF-β1刺激通过NLRP3增加线粒体ROS。
维生素A和脂质代谢--视黄醇储存和脂滴
维生素A稳态的调节是HSCs在健康和损伤肝脏中的一个本质特征。HSCs储存50%-95%的人体维生素A,由视黄醇及其代谢物组成。超过95%的类视黄醇以视黄酯(REs)形式储存,占HSC细胞质脂滴含量的30%-50%。在HSCs中,类视黄醇与几种脂滴蛋白有关,包括脂肪分化相关蛋白(ADRP)、脂肪素、perilipin 5、ATGL和CGI-58,以及肝脂肪酸结合蛋白(L-FABP)。一般来说,更高的perilipin表达降低了HSC的激活,可能是通过稳定类视黄酯液滴来减弱它们对提供能量的脂肪酸的分解代谢。
血液维生素A水平受到严格的调控,但健康肝脏中维生素A摄取和释放的具体机制尚未完全阐明。HSCs对视黄醇的摄取既以游离视黄醇的形式,也以holo-RBP、视黄醇结合蛋白(RBP)和视黄醇的结合形式,后者优先酯化。HSCs的类视黄醇含量也是异质的。原发性HSCs的差异FACS分类(视黄醇自身荧光的分类与胶原促进因子驱动的GFP的表达)揭示了健康肝脏中至少两个主要亚群,前者是具有较高视黄醇储存能力的亚群体,而后者是具有更加MFB样表型的亚群体,表明这些HSCs可能是“启动”了的,以响应肝脏损伤。在“启动”HSCs中,与视黄醇分解代谢有关的基因的上调和较小的脂滴揭示了HSC脂质代谢的一些早期变化,这些变化可能发生在体内肝脏损伤情况下。维生素A处理HSCs可以抑制培养活化,并能部分维持qHSC标记的表达,而抑制MFB标记的表达。此外,视黄醇暴露也会导致aHSC部分恢复到静息状态。
HSC激活过程中视黄酯水解酶(REHs)从脂滴中释放REs,有许多潜在的酶密切相关,包括脂肪甘油三酯脂肪酶(ATGL/PNPLA2)、PNPLA3、激素敏感脂肪酶和羧基酯脂肪酶。体外培养研究表明,HSCs释放视黄醇与RBP或结合蛋白无关,因为抑制蛋白质分泌不能调控视黄醇的分泌。然而,视黄醇分泌的主要方式是HSCs直接将游离视黄醇释放到血液中还是肝细胞分泌holo-RBP仍有争议。HSC的激活似乎需要通过自噬丢失脂滴,这可能是促进这种代谢要求的细胞反应的关键(Figure 3)。HSC的激活增加了细胞内的REH活性,并随后引起视黄醇的释放,而不增加REs的释放。aHSCs细胞外REH活性的缺乏进一步意味着细胞内水解介导视黄醇丢失。在增加水解酶活性的同时,aHSCs也降低了视黄醇酯化的能力,因为HSC的激活导致LRAT的表达迅速消失。
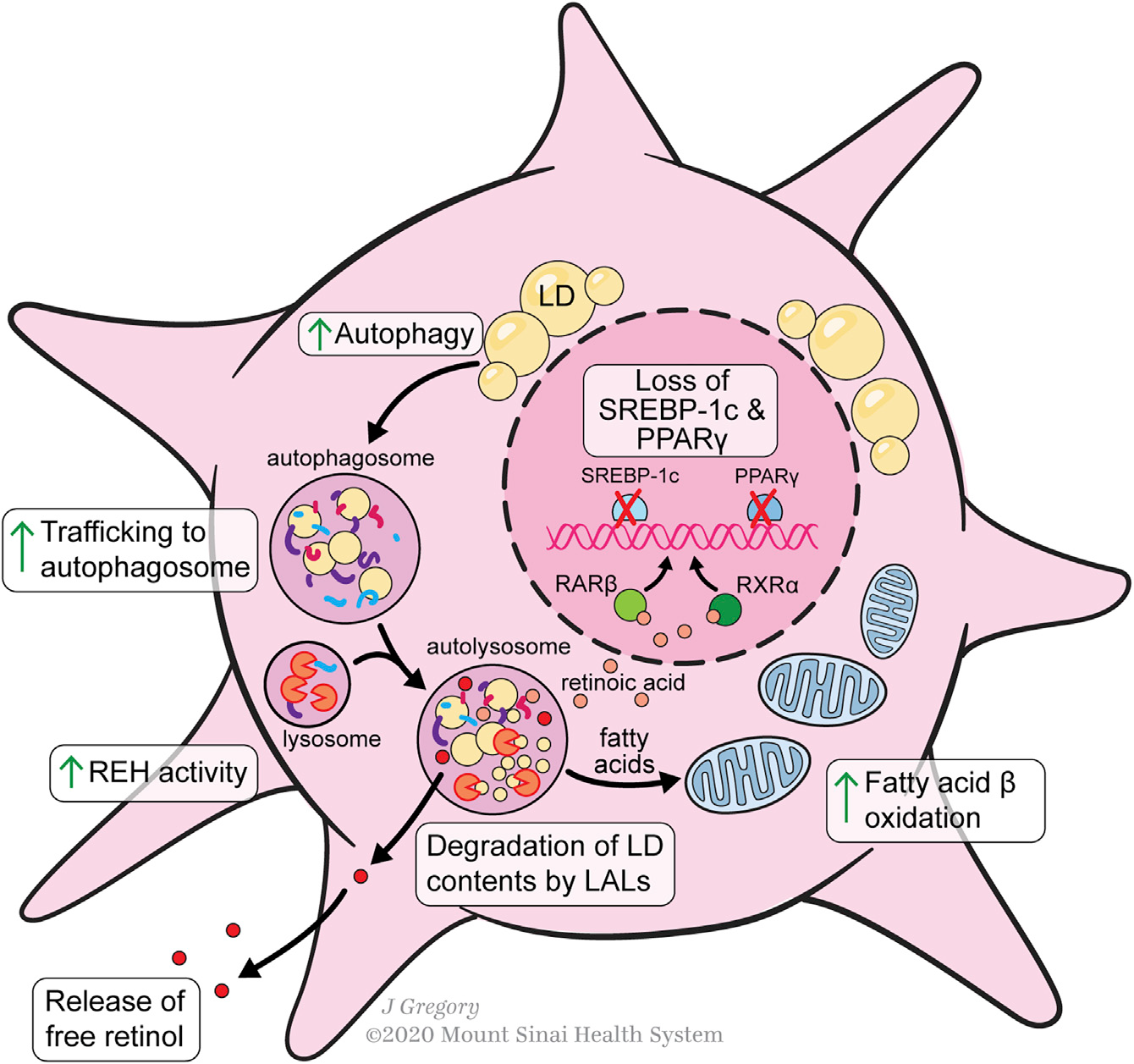
Figure 3. Lipid Metabolism during HSC Activation
成脂表型和脂肪酸
脂肪酸含量和代谢的调节因子控制HSC的激活。过氧化物酶体增殖物激活受体γ(PPAR-γ)和甾醇调节-元素结合蛋白-1(SREBP-1c)是HSC静息的标志, PPAR-γ和SREBP-1c的异位诱导可以逆转HSC的激活。脂肪酸为视黄醇在qHSCs中的酯化提供了重要的底物。
HSC激活过程中脂滴被代谢,为氧化提供脂肪酸类物质,在已知的脂肪酸氧化激活剂PPAR-β的控制下产生活化/转分化的能量。PPAR-γ和SREBP-1c下调是通过激活MAPK/ERK信号级联来实现的,几种促纤维化细胞因子之间存在关联。
在活化的早期阶段,最丰富的脂肪酸-棕榈酸、油酸、棕榈酸和硬脂酸-达到峰值浓度。活化后期游离脂肪酸总量下降,而花生四烯酸(C20:4)和二十二碳六烯酸(C22:6)相对富集,以及脂肪酸伸长酶(Elovl5, Elovl6)和脱饱和酶(Scd1)上调。HSC脂肪酸含量的总体下降与成脂表型的丧失是一致的。脂肪酸β-氧化是HSCs激活的重要能量来源,因为线粒体脂肪酸分解代谢的抑制阻碍了HSC的激活。
乙酰辅酶A羧化酶(ACC)在HSC激活过程中参与了代谢重编程。通过降低αSMA的表达和胶原的产生,ACC抑制降低了HSC的激活。ACC抑制剂的抗纤维化活性是通过抑制DNL,这是在HSC激活期间诱导糖酵解和氧化磷酸化所必需的。虽然DNL调节HSC代谢的分子机制尚未阐明,但ACC抑制被认为是NASH的一种治疗方法,因为它既降低了肝细胞的脂毒性,又抑制了HSC的纤维发生,但ACC在体内的抑制作用尚未直接归因于其对HSCs的影响。
胆固醇
胆固醇有助于NASH纤维发生的发病机制,因为HSC的激活可以通过游离胆固醇(FC)的积累来诱导。在培养的HSCs中,FC的积累增加了Toll样受体4(TLR4)的水平,致使HSCs对TGF-β1诱导的激活敏感。HSCs中FC的积累由低密度脂蛋白受体(LDLR)和miR-33a(胆固醇代谢的microRNA调节剂)控制,两者都在HSC激活过程中将其上调。
谷氨酰胺分解
ECM生成的巨大代谢需求不仅通过碳水化合物和脂质代谢重编程,还通过蛋白质代谢来满足。比较qHSCs和aHSCs之间差异表达的代谢基因,只有6%的这类基因涉及碳水化合物代谢,38%参与蛋白质代谢,这表现为向谷氨酰胺分解的转变。谷氨酰胺分解是将谷氨酰胺转化为α-酮戊二酸,通常发生在癌细胞,活化的HSCs上调了谷氨酰胺酶GLS1的表达水平。
HSCs的谷氨酰胺剥夺可引起脂质积累,增加PPAR-γ的表达,降低胶原蛋白I的表达,这表明谷氨酰胺分解是aHSC的重要能量来源。在体内,HSCs对谷氨酰胺分解标志着活跃的纤维化发生,其细胞特异性拮抗作用通过剥夺细胞中的谷氨酰胺而成为潜在的治疗靶点。与谷氨酰胺相比,葡萄糖剥夺没有抗纤维化作用,尽管aHSCs增加了糖酵解基因的表达和葡萄糖的消耗。
HSC代谢与应激反应的联系
氧化应激ROS
产生ROS是aHSC的重要特征,因为敲除或抑制产生ROS的酶会抑制HSC的激活。ROS产生的增强与线粒体活性的增加相一致。
几种典型的HSC激活因子集中于ROS介导的激活,特别是通过NADPH氧化酶的上调(Figure 4)。ROS的产生被认为是纤维化细胞活化的“前馈”机制,特别是通过TGF-β1。氧化还原失衡激活TGF-β1的潜在形式,而TGF-β1信号则产生氧化还原失衡。TGF-β1信号通路上调了几个NADPH氧化酶-- NOX1、NOX2、NOX4和NOX5,H2O2生成增加,从而提高了胶原的产量。
在胚胎、肾脏和心脏成纤维细胞中进行的研究表明,线粒体ROS的产生增加。此外,线粒体ROS由锰超氧化物歧化酶(Sod2)代谢到H2O2可上调胚胎成纤维细胞基质金属蛋白酶的转录,增加Sod2的活性和MMP-1、MMP-2、MMP-3和MMP-7的表达。相反,内源性大麻素介导的线粒体ROS的增加导致HSC细胞死亡,这与线粒体ROS作为细胞凋亡调节因子的经典作用是一致的。这两个主要的ROS产生轴的差异调节可能解释了线粒体ROS更可变的贡献,而不是NOX酶的最终促进纤维化。
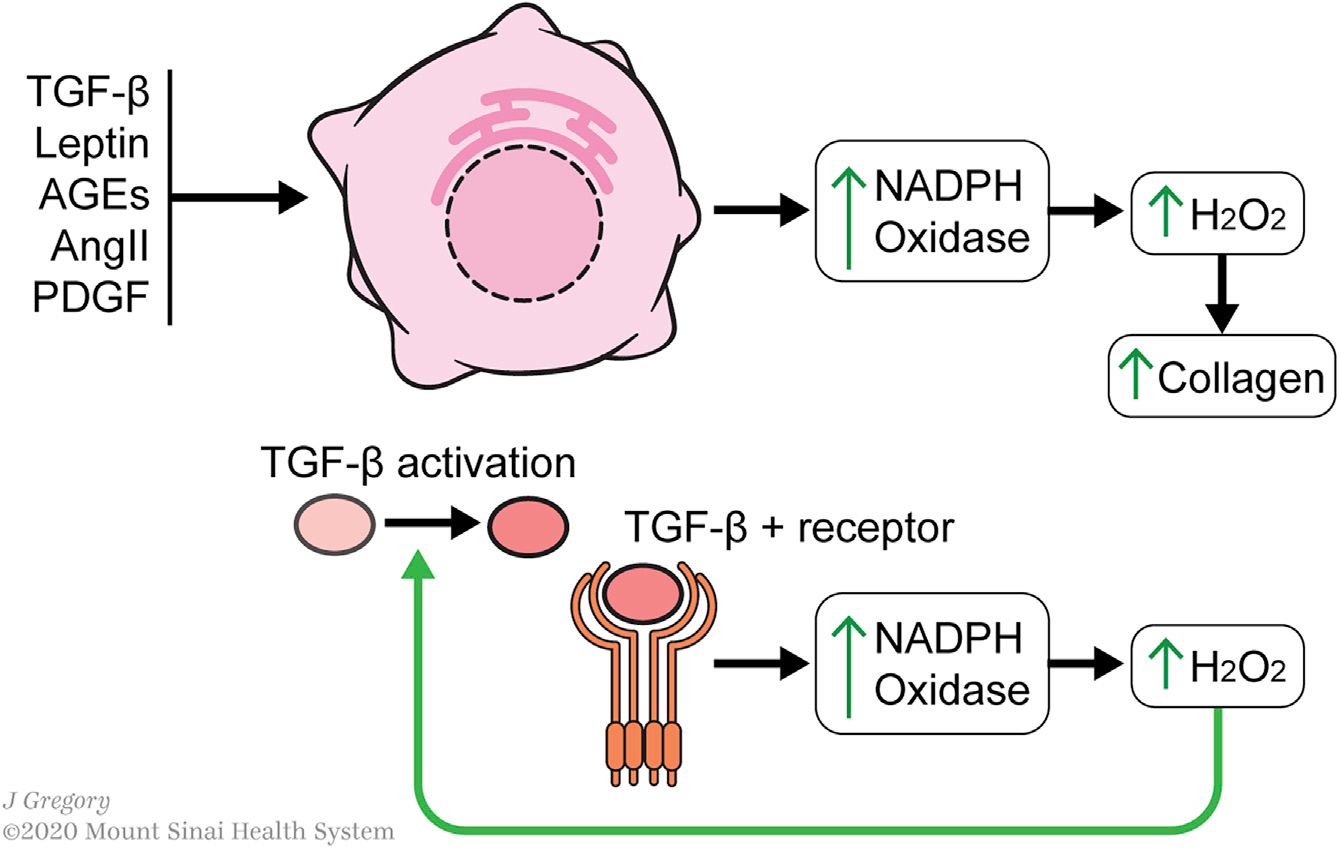
Figure 4. Redox Metabolism in HSC Activation
抗氧化反应
HSCs的内源性抗氧化活性没有得到很好的表征。在体外,TGF-β1激活的HSCs增加了谷胱甘肽含量,这与培养激活的HSCs的研究是一致的。此外,不同程度的HSC激活对应于不同的氧化还原状态。在一项利用M1-4HSC细胞系的晚期细胞激活的研究中,TGF-β1促进进一步激活,导致肌成纤维M-HT细胞完全转分化。在培养中,M1-4HSCs含有较高的细胞内ROS含量,而NOX活性较低。完全转分化的M-HTs提高了Sod2的活性和谷胱甘肽水平,这可能是抑制ROS诱导的细胞损伤所必需的。此外,增加抗氧化可用性可减轻促纤维化信号如TGF-β1和PDGF的影响,并减少疾病动物模型中的纤维化。
自噬
大自噬(“自噬”)是一种细胞应激反应,参与大分子和细胞器的自溶酶体消化,产生细胞内营养和能量。上述几种与激活相关的代谢变化集中于自噬细胞,包括脂解的增加、ROS的产生和ER应激。脂滴运动是HSC激活的一个关键特征,是由自噬驱动的。早期研究脂滴运动的影响集中在细胞内视黄酯水解后向细胞外释放游离视黄醇,目前已知水解的一个关键结果是释放游离脂肪酸为细胞激活“供能”。因此,添加内源性油酸可以挽救自噬缺陷的HSCs的激活,而抑制脂肪酸氧化则具有相反的作用。另一方面,由于缺乏卵磷脂-视黄醇酰基转移酶(LRAT),缺乏脂滴的小鼠HSC仍然可以完全激活,但不会自发激活。
非细胞自主作用的HSC代谢
肝星状细胞-肝细胞代谢串扰
肝细胞占肝脏内细胞的80%以上,通过交换类视黄醇代谢物和信号方式与HSC代谢相互作用。在HSC激活过程中,HSCs释放的游离视黄醇被肝细胞以接触依赖的方式迅速吸收,以holo-RBP的形式进入循环。除了在HSC激活过程中失去类视黄醇,目前尚不清楚维生素A的肝脏代谢是否直接导致肝损伤和纤维化。视黄醇在肝脏中总含量随人类肝病的进展而下降,在两种NASH小鼠模型中视黄醇-RBP4耗竭,视黄醇重新分布到肝细胞而不是HSCs。
Hh(Hedgehog)下游的信号通路和瘦素受体结合调节HSCs中的葡萄糖代谢。Hh信号广泛参与伤口愈合,Hh配体是肝细胞在肝脏损伤过程中产生的。这些配体作用于邻近的HSCs,通过HIF-1α转录因子诱导激活,HIF-1α转录因子是能量代谢的主调节因子,可激活大量的靶基因,包括葡萄糖转运蛋白和糖酵解酶等。
HSCs作为免疫代谢的影响因素
虽然免疫代谢的新概念主要集中在免疫细胞,但HSCs也显著促进了metainflammation和组织损伤的收敛途径,特别是在酒精性和非酒精性脂肪性肝炎(NASH)中。通过控制类视黄醇代谢,它们调节肝脏和全身免疫中的免疫细胞功能。星状细胞产生炎症介质的反应,包括细胞因子和趋化因子,可以放大肝脏对损伤的反应。
HSCs也被认为是抗原提呈细胞,通过Toll样受体信号和炎症小体激活对天然免疫配体作出反应。HSCs与炎症细胞的相互作用可通过旁分泌信号驱动炎症和损伤。例如,HSC通过MER-TK受体与巨噬细胞的相互作用有助于实验性NASH的纤维化,以及与NK、NKT和γδT细胞的串扰。
有几个例子值得注意:(1)HSCs释放全反式RA通过诱导精氨酸酶-1和诱导型一氧化氮合酶促进树突状细胞的耐受性表型;(2)通过诱导免疫调节分子吲哚胺2,3-双加氧酶1(IDO1),诱导脂多糖(LPS)对FoxP3+调节性T细胞的增殖,该酶分解色氨酸生成犬尿氨酸。而抑制IDO可以减少培养的HSCs的增殖。(3)不对称二甲基精氨酸(ADMA)被二甲基氨基水解酶(DDAH)分解代谢,降低了TGFB1介导的原代大鼠HSCs的活化;(4)细菌产物微囊藻毒素-亮氨酸精氨酸酶(MC-LR)可通过Hh信号激活HSCs,给药可诱导体内肝纤维化。
小结
虽然基于HSCs代谢调节的治疗迄今为止是有限的,但阐明其代谢途径为治疗肝病和与肝脏有关的系统性疾病提供了一个的新的潜在靶点。目前一些针对减轻纤维化的代谢途径的药物正在进行研究中,包括:(1)胆汁酸,具有抗凋亡和减轻ER应激的特性;(2)FXR激动剂,调节葡萄糖、脂质和胆汁酸的代谢;(3)甲状腺激素受体-β激动剂可调节脂质和葡萄糖稳态;(4)PPAR激动剂,成脂的主要调节剂;(5)维生素A偶联用于HSC靶向。
HSCs参与调节碳水化合物、线粒体、脂质和类视黄醇稳态的途径,在健康和疾病中发挥重要作用。在慢性肝损伤中,HSCs驱动肝纤维化,并与炎症和癌症有关。细胞从静息状态激活或转分化,进入增殖的、运动的肌成纤维细胞,分泌细胞外基质,这需要快速适应,包括中心碳代谢重编程、线粒体数量和活性的增强、内质网应激,以及通过自噬依赖的水解细胞质液滴中的视黄酯释放游离脂肪酸,以满足更高的能量需求。作为其他组织周皮细胞的原型,对HSC代谢驱动因素和弱点的识别具有针对这些途径进行治疗以增强实质生长和调节修复的潜力。
参考文献
Parth Trivedi, et al. The Power of Plasticity—Metabolic Regulation of Hepatic Stellate Cells. Cell Metabolism . 2021.
原文阅读,请长按识别下方二维码

精彩推荐
3. Cell Metabolism | 肠道菌群相关代谢物--胆汁酸生物学功能
4. Protein&Cell | 胆汁酸替代合成途径与代谢性疾病
5. 肠·道 | 贾伟:肝病肠治?且看肠道细菌如何对肝病推波助澜!
6. Cell子刊 | 癌细胞并不孤单--肿瘤微环境中的代谢通信
7. CRIT REV FOOD SCI︱肠道微生物群与脂溶性维生素
